
May 02, 2024
【Case study】Target Analysis
Structure modeling of human aryl hydrocarbon receptor using protein AI and MD simulation and structure prediction
In recent years, AlphaFold2 (AF2), *1 a protein three-dimensional structure prediction program, has received considerable attention. We have been utilizing this technology for modeling protein structures for use in docking simulations in Structure-Based Virtual Screening (SBVS). In this article, we introduce a case study on the use of protein conformation prediction software in SBVS for the human aryl hydrocarbon receptor (AhR), which we previously introduced as a case study.*2
1. Structure prediction of human AhR by protein structure prediction AI
AF2 is a protein structure prediction program based on deep learning technology published by DeepMind, Inc. that can predict protein structures from their amino acid sequences with high accuracy. AF2 is limited in that it can only generate structures in the apo state (ligand unbound state) due to the characteristics of the structural data set used to build the prediction model. Recently, however, it has been shown that by changing the depth of Multiple-Sequence Alignment (MSA) for multiple amino acid sequences in AF2 structure prediction, multiple different 3D structures can be generated, and in some cases it is possible to generate structures close to the holo state (ligand bound state).*3
ColabFold*4 has been proposed as an Open Source Software (OSS) that makes AF2 easier to use for general users, and the analysis in this article uses LocalColabFold,*5 a local version of this software (hereinafter referred to as “ColabFold”).
1.1. Structure prediction by ColabFold
There are multiple structural prediction parameters for ColabFold, and they can be combined in a wide variety of combinations. However, the optimal combination of these prediction parameters is not clear, perhaps because it varies from protein to protein. However, for a protein like AhR, which has only about 100 amino acid residues, a single structure prediction by ColabFold takes only a few tens of seconds using NVIDIA’s Tesla V100 GPU-equipped computer. Therefore, for this validation, we decided to generate a large number of predicted structures under various combinations of prediction parameters (note that for larger proteins, the prediction computation time increases exponentially as the number of residues increases). For each of these predicted structures, we evaluated the performance of the antagonist predictions by docking simulations on known antagonist, agonist, and decoy datasets for human AhR and employed an approach to find the optimal structure in SBVS (Figure 1.1).
The input information to ColabFold is the same amino acid sequence used in the homology modeling of human AhR in our case study.*2 ColabFold can also use a template structure for structure prediction. Therefore, in this validation study, whether template structures (apo and holo structures) were used or not was treated as one of the prediction parameters. In the end, we generated approximately 1,000 predicted human AhR structures.
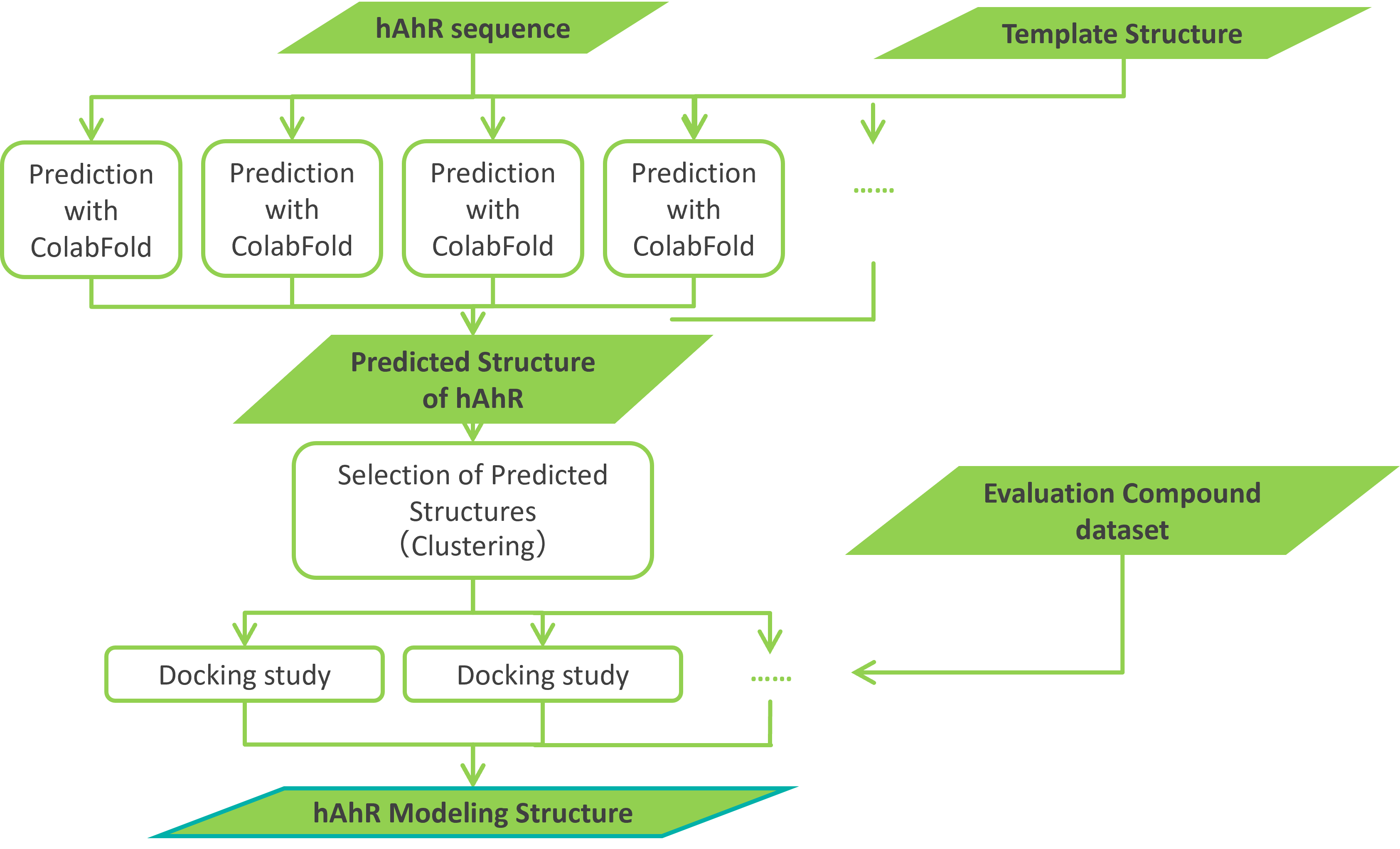
Figure 1.1. Procedure for modeling the structure of human AhR using ColabFold.
1.2. Selection of generated predicted structures
Next, the prediction performance of these generated structures for identifying antagonist compounds was evaluated by docking simulations. However, since there were many structures to be evaluated, similar structures were clustered in advance to reduce computational cost, and a total of 16 structures were selected as representative structures for each cluster.
1.3. Evaluation of predicted structures by docking simulation
As in our previous case study,*2 we evaluated the prediction performance of each structure by docking simulation using two known compound datasets: (1) antagonist vs. agonist and (2) antagonist vs. decoy to obtain a human AhR structure that can discriminate between antagonist and decoy. The prediction performance of each structure was evaluated by docking simulations.
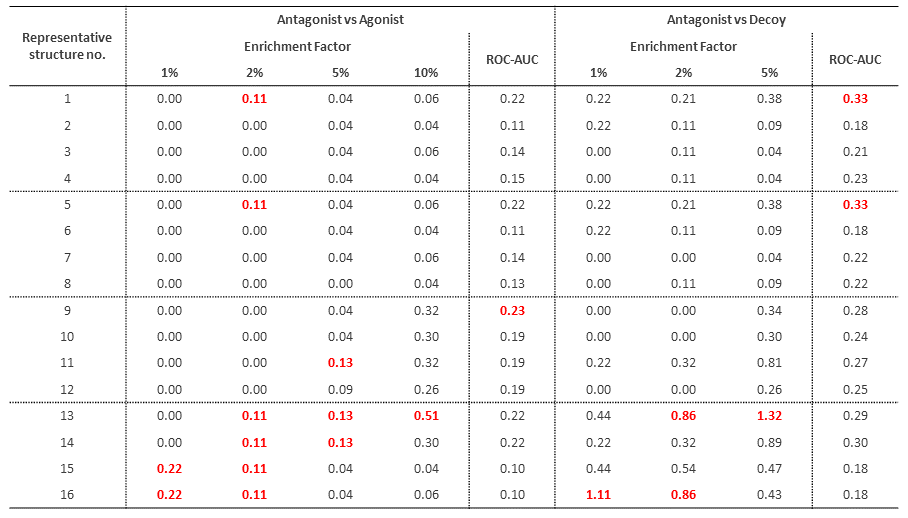
The results are shown in Table 1.1, where the Enrichment Factor is the ratio of the enrichment of active compounds by screening, and the Enrichment Factor $EF_{x\%}$ when $N_{tot}$ compound sets are screened and $N(x\%)$ compounds with the top $x\%$ scores are obtained is $\frac{N_{active}(x\%)⁄N(x\%)}{N_{active}⁄N_{tot}}$ (where $N_{active}(x\%)$ is the number of active compounds in the top $x\%$ of scores and $N_{active}$ is the total number of active compounds). If $EF_{x\%}$ exceeds 1, it means that active compounds were enriched by screening. ROC-AUC is the area under the ROC curve, and the closer the value is to 1, the better the classification model is (0.5 for random classification). As indicated by the $EF_{x\%}$ and ROC-AUC for each structure, the prediction performance was generally poor for all structures, although the use of holo template structures (No. 13-16) shows a relatively better trend. This is because the molecular weight of the bound antagonist ligand in the holo template co-crystal structure is smaller (see Figure 1 in our case study*2), and the shape of the binding pocket is not much different from that of the apo template structure.
Table 1.1. Docking evaluation results for 16 representative structures against the test compound set.
2. Structure optimization by MD simulation
We next attempted to improve the predictive performance of the generated ColabFold structure by structure optimization using Molecular Dynamics (MD) simulations (Figure 2.1).
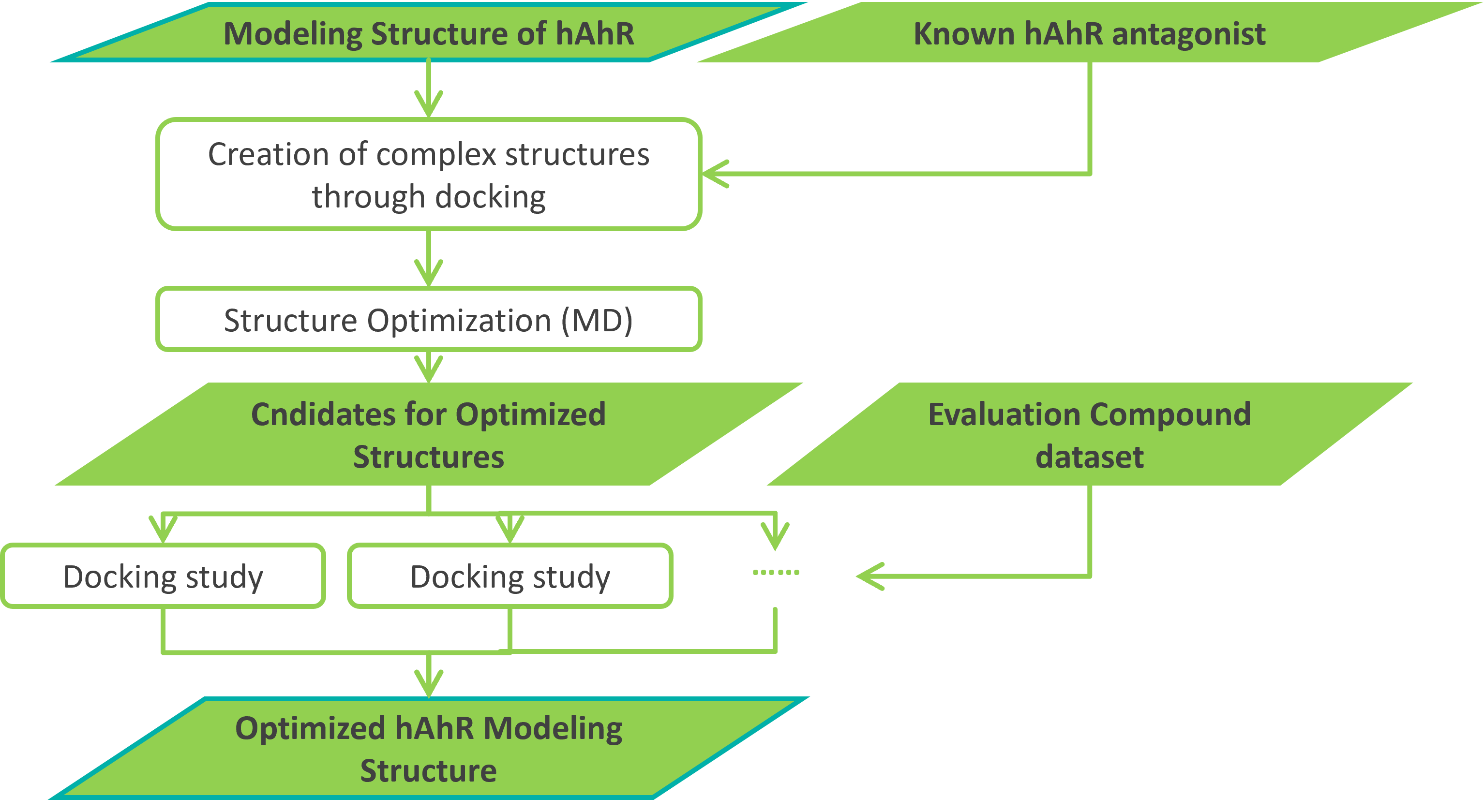
Figure 2.1. Structure optimization flow for MD simulation of human AhR structure prediction.
2.1. Structural optimization by MD
There are various approaches to optimize the ColabFold predicted structure, but we mainly use MD simulations. However, while ordinary (classical) MD simulations have the advantage of observing dynamics, such as the response to arbitrary inputs such as ligand binding or charge state changes, they require a large amount of simulation time to generate a sufficient number of rare structural transitions when sampling a wide range of structures. Thus, classical MD simulations are not well suited for the structural optimization of macromolecules, such as proteins. To solve this problem, various methods have been proposed to improve the efficiency of structure sampling, collectively known as extended ensemble methods,*6 among which the Accelerated MD (aMD) method*7 is employed in this verification study (unless otherwise noted, references below to “MD simulation” employed this method) (Figure 2.2).
In some extended ensemble methods, the direction (reaction coordinates) of an arbitrary reaction to be observed, such as a conformational change or a binding/dissociation reaction of molecules, is defined in advance, and random walks in that direction on a free energy surface are performed to make structural sampling more efficient. However, determining the reaction coordinates is not easy in general due to the fact that reaction coordinates vary depending on the simulation system and the observation target, and it is not clear whether the reaction coordinates truly represent the reaction or not. On the other hand, the aMD method is an extended ensemble method that does not require reaction coordinates and is relatively easy to use.
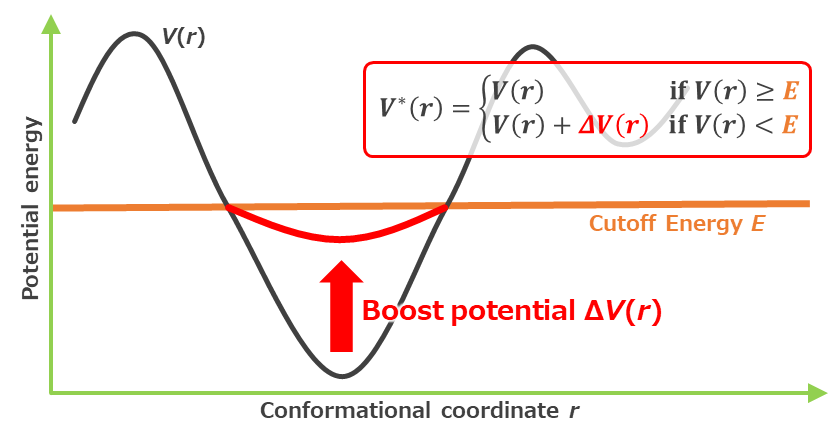
Figure 2.2. Schematic of the Accelerated MD (aMD) method.*7 When the potential energy $V(r)$ of the system is lower than the threshold $E$, a boost potential ${\Delta}V(r)$ is applied to smooth the potential energy surface of the system to improve the sampling efficiency.
In this study, we adopted the approach of creating a bound complex structure with a representative antagonist selected by us for the 16 representative human AhR cluster structures created in §1, and performing MD simulations. Analysis of the snapshot structures obtained from the latter part of the MD simulation shows that the sampled structures are different from the known crystal structures (apo and holo states) of AhR (which were also used as template structures for ColabFold) (Fig. 2.2). (Figure 2.3). This result suggests that there are roughly five metastable states.
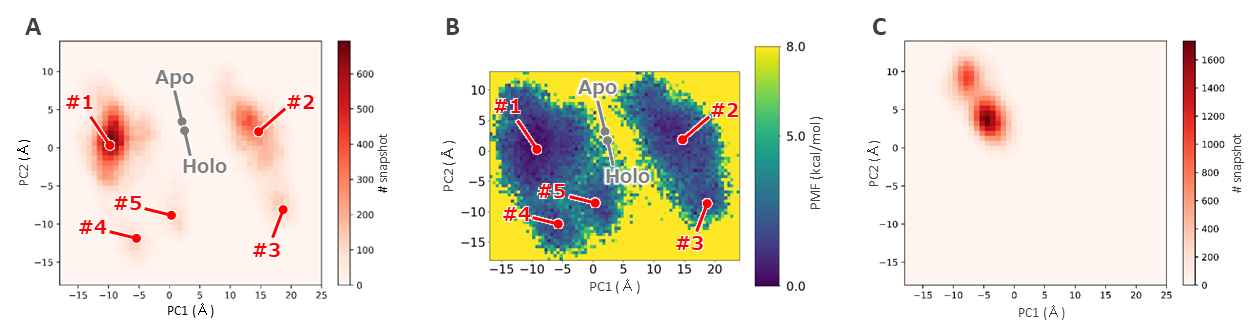
Figure 2.3. Distribution of snapshot structures from MD simulations. (A) Two-dimensional histogram, (B) Performance Measurement Framework (PMF): x and y axes are the first and second principal component axes, respectively. (C) Distribution of the snapshot structure obtained from a classical MD simulation of the same time. Compared to the MD simulation method employed in this verification study, only a portion of the structural states are sampled.
2.2. Evaluation of optimized structure by docking simulation
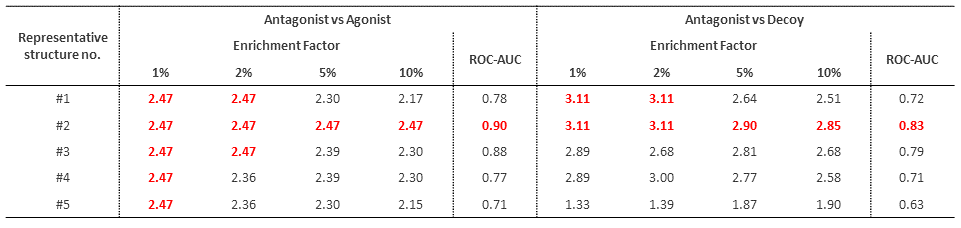
First, we grouped the snapshot structures from the MD simulations into five clusters and evaluated the prediction performance for antagonists by docking simulations similar to §1.3 for each of the representative structures in each cluster. The results show a significant improvement in prediction performance for all structures (Table 2.1). In particular, the ROC-AUC of the structure with the best prediction performance (#2) is higher than that of the structures obtained by combining conventional homology modeling and classical MD simulations (antagonist vs agonist: 0.80, antagonist vs decoy: 0.79 ).*2
Table 2.1. Docking evaluation results for five structures obtained from MD simulations
3. Summary and issues
In this article, we introduced a case study of structure modeling using a combination of protein structure prediction software and structure optimization by MD simulation for the SBVS search of antagonists of human AhR, which was introduced in our previous case study.*2 In conclusion, it was difficult to use the predicted structure from ColabFold for human AhR as is for SBVS due to issues with the reported crystal structure (§1.3), but by introducing structure optimization by MD simulation, we were able to obtain a structure model with sufficient predictive performance to enable SBVS studies.
It should be noted that the performance of antagonist prediction by the structure model of human AhR in this validation study is not significantly different from that obtained by the combination of conventional homology modeling and MD simulation, but there is a significant difference in the amount of effort required to obtain the structure model. In SBVS, the accuracy of the entire flow is greatly affected by the quality of the target protein structure, so even if the a crystal structure is used as is, the structure must be prepared with great care. In the case of conventional homology modeling, it is necessary to carefully consider many factors such as sequence similarity between the amino acid sequence to be modeled and the amino acid sequence of the template structure, resolution of the structure, and experimental conditions. However, these factors currently require a large amount of manual work, which inevitably results in a considerable labor and associated cost. On the other hand, in this validation study, ColabFold was used as the protein structure prediction program, which made it possible to replace a large part of the manual labor with ColabFold, resulting in a significant reduction (about 75%) of the work required for the structure prediction of the human AhR in §1. Currently, a variety of new protein structure prediction methods and software are currently being proposed in the literature, and their successful use (after carefully examination of their strengths and weaknesses) may lead to a significant improvement in the efficiency of computational work.
However, the approach presented in this article also has its challenges. First, (a) regarding the use of ColabFold, we generated several different predicted structures of human AhR by changing the structure prediction parameters of ColabFold, but whether this approach is generally effective (e.g., whether it can be changed while maintaining the correct structure, how diverse the structures generated can be, etc.) needs to be tested further. Second, (b) the MD simulation method used in this study has some advantages over other extended ensemble methods, but the results obtained are still arbitrary due to the existence of arbitrary parameters such as the energy threshold $E$ and simulation time. Since the appropriate structural optimization method may differ depending on the situation, the advantages and disadvantages of each extended ensemble method should be well understood and considered before using an approach based on MD simulations. Finally, (c) regarding the prediction of protein conformation in the holo state, there are recently announced structure prediction programs that directly predict the protein-ligand complex structure,*8,*9 and it may be worth considering the use of these new tools. (Even if the complex structure is predicted using these tools, it may not be possible to predict the structure of the protein-ligand complex using MD simulations).
In conclusion
Xeureka supports your drug discovery research, including stand-alone requests such as this for Target Analysis, as well as requests involving Hit Identification, Hit to Lead, and Lead Optimization. We are happy to provide free of charge proposals including feasibility studies for our in silico approaches. We are also available for consultation focused on tailored solutions or processes such as protein structure generation or the construction of ADME prediction models. Please contact us from the contact page below!
CONTACT | Xeureka Inc. (xeureka.co.jp)
References
- *1: Jumper, et al. (2021) Nature 596:583–589.
- *2: Xeureka Inc., 【Case Study】Ultra Large Scale Virtual Screening
- *3: del Alamo, et al. (2022) eLife 11:e75751.
- *4: Mirdita, et al. (2022) Nature Methods 19:679-682.
- *5: LocalColabFold, https://github.com/YoshitakaMo/localcolabfold
- *6: Okamoto (2004) J. Mol. Graphics Model. 22:425-439.
- *7: Hamelberg, Mongan, and McCammon (2004) J. Chem. Phys. 120:11919-11929.
- *8: Lu, et al. (2024) Nat. Commun. 15:1071
- *9: Plainer, et al. (2024) OpenReview, https://openreview.net/forum?id=1IaoWBqB6K