
May 02, 2024
【事例紹介】Target Analysis
タンパク質構造予測AIとMDシミュレーションによるヒトAryl hydrocarbon receptorの構造モデリング
近年、タンパク質立体構造予測ソフトウェアであるAlphaFold2 (AF2)*1が脚光を浴びています。弊社でもその技術をStructure-Based Virtual Screening (SBVS)におけるドッキングシミュレーションなどで利用するタンパク質の立体構造のモデリングなどで活用しています。本記事では弊社事例紹介*2でも紹介したヒトAryl hydrocarbon receptor (AhR)を対象に、SBVSにおいてタンパク質立体構造予測ソフトウェアを使用した事例についてご紹介します。
1. タンパク質構造予測AIによるヒトAhRの構造予測
AF2はDeepMind社が発表したディープラーニング (深層学習)技術を用いたタンパク質立体構造予測ソフトウェアで、タンパク質のアミノ酸配列から高い精度でその立体構造を予測することが出来ます。通常AF2はその予測モデル構築時に使用した構造データセットの特徴から、基本的にはapo状態 (リガンドが非結合の状態)の立体構造しか生成できないという限界がありました。しかし最近、AF2の構造予測において多重アミノ酸配列アライメント (Multiple-Sequence Alignment, MSA)の深度を変化させることにより複数の異なる立体構造を生成することができ、場合によってはholo状態 (リガンドが結合した状態)に近い構造の生成も可能であることが示されています*3。
このAF2を一般ユーザー向けに使いやすくしたOpen Source Software(OSS)としてColabFold*4が提案されており、本記事の解析においてはこれローカル版に当たるLocalCorabFold*5を利用しています (以後「ColabFold」と略記します)。
1.1. ColabFoldによる構造予測
ColabFoldの構造予測パラメータは複数存在し、その組み合わせは多岐に及びます。しかしこれら予測パラメータの最適な組み合わせは、おそらく予測タンパク質によって変化するために明らかではありません。ですがAhRのような100アミノ酸残基程度のタンパク質の場合、ColabFoldによる一回の構造予測にはNVIDIA社のTesla V100 GPU搭載コンピューターを使用すると数十秒程度の時間しか要しません。そこで本検証では、様々な予測パラメータの組み合わせの下で多数の予測構造を生成することにしました(ただしより大きなタンパク質では、その残基数が増えるに伴って爆発的に予測計算時間が増えますのでご注意ください)。この予測構造それぞれについてヒトAhRに対する既知antagonist, agonistおよびdecoyデータセットを用いたドッキングシミュレーションによりantagonist予測性能を評価し、SBVSにおける最適な構造を見つけ出すアプローチを採用しました (図1.1)。
ColabFoldへの入力情報には、弊社事例紹介*2におけるヒトAhRのホモロジーモデリングにおいて使用したものと同一のアミノ酸配列を使用しました。またColabFoldは構造予測の鋳型 (テンプレート) となる構造を使用することも可能です。そこで本検証では、テンプレート構造 (apo構造およびholo構造) の使用の有無についても予測パラメータの一つとして扱いました。最終的に約1,000個のヒトAhR予測構造を生成しました。

図1.1. ColabFoldを用いたヒトAhRの構造モデリング手順
1.2. 生成した予測構造の選抜
次にこれら生成構造のantagonist予測性能をドッキングシミュレーションにより評価します。しかし現状では評価対象の構造が多数あるため、計算コスト削減のためにこのドッキング評価に先立って予め構造クラスタリングを行い、各クラスターの代表構造として計16構造を選抜しました。
1.3. ドッキングシミュレーションによる予測構造の評価
弊社紹介事例*2と同様、antagonistを識別できるヒトAhR構造を取得するために(1) antagonist対agonist、(2) antagonist対decoyの2通りの既知化合物データセットを使用して、ドッキングシミュレーションにより各構造の予測性能を評価しました。
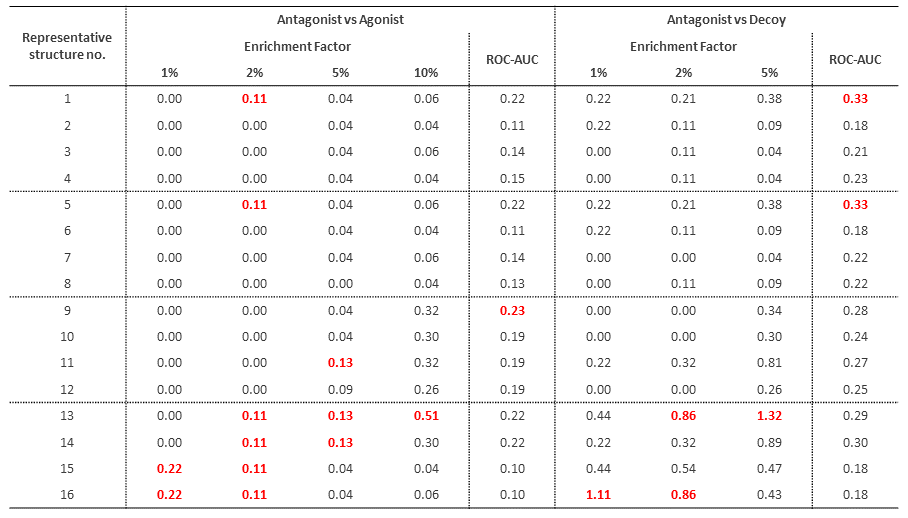
結果は表1.1の通りです。ここでEnrichment Factorはスクリーニングによる活性化合物の濃縮度を表す比率で、$N_{tot}$個の化合物セットをスクリーニングして$N(x\%)$個のスコア上位$x\%$の化合物を取得した場合のEnrichment Factor $EF_{x\%}$は$\frac{N_{active}(x\%)⁄N(x\%)}{N_{active}⁄N_{tot}}$で表されます (ここで$N_{active}(x\%)$はスコア上位$x\%$に含まれる活性化合物数で、$N_{active}$は全活性化合物数)。$EF_{x\%}$が1を超えるとスクリーニングにより活性化合物が濃縮されたことを意味します。またROC-AUCはROC曲線下面積であり、値が1に近いほど分類モデルとして優れていることを意味します (0.5: ランダム分類時)。各構造の$EF_{x\%}$とROC-AUCが示す通り、holoテンプレート構造を使用した場合 (No. 13-16)が相対的に良好な傾向を示すものの、概してどの構造も予測性能が不良でした。これはholoテンプレート構造における共結晶antagonistリガンドの分子量が小さく (弊社事例紹介*2における図1を参照下さい。全体としてantagonistの方がagonistよりも分子量が大きい傾向があります)、そのbinding pocket形状がapoテンプレート構造とあまり変わらないことに起因するものと推測されます。
表1.1. テスト化合物セットに対する16代表構造のドッキング評価結果
2. MDシミュレーションによる構造最適化
続いてColabFoldの生成構造の予測性能を分子動力学 (Molecular Dynamics, MD)シミュレーションを用いた構造最適化により改善することを試みました (図2.1)。
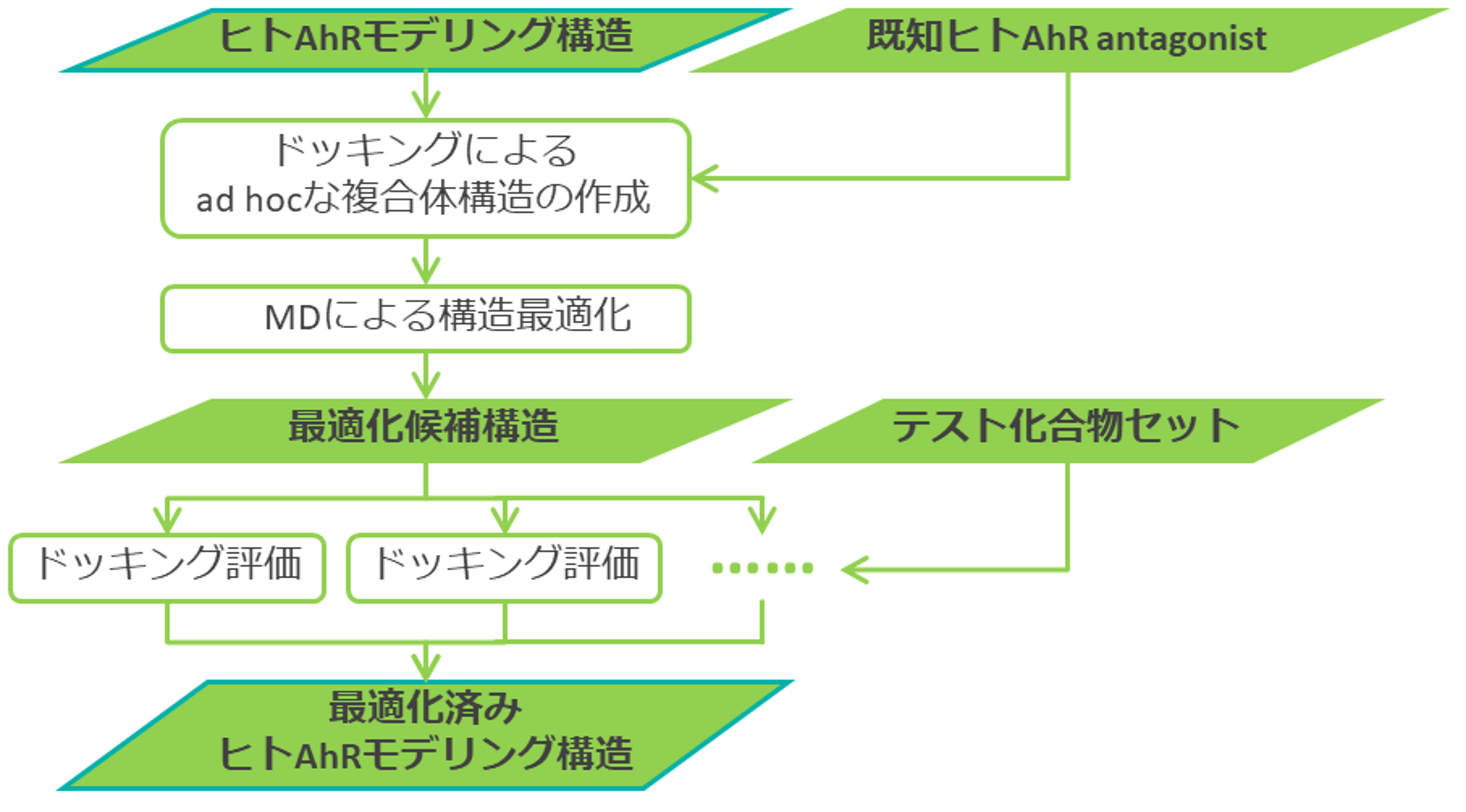
図2.1. ヒトAhR予測構造のMDシミュレーションによる構造最適化フロー
2.1. MDによる構造最適化
ColabFoldの予測構造の最適化には様々なアプローチ方法が考えられますが、弊社では主にMDシミュレーションによるアプローチを採っています。しかし通常の (classicalな)MDシミュレーションはどちらかと言えば、リガンド分子の結合や荷電状態変化などの任意の入力に対するレスポンスを見るなどのダイナミクス観察に強みがあり、広範な構造のサンプリングにおいてはレアイベントである構造遷移を十分な回数生じさせるために多大なシミュレーション時間を必要とすることから、タンパク質のような巨大分子の構造最適にはclassicalなMDシミュレーションはあまり向いていないと言えます。この問題を解決するべく、拡張アンサンブル法と総称される構造サンプリングを効率化する様々な手法が提案されてきました*6。本検証では、その中でもAccelerated MD (aMD)法*7を改良した手法を採用しています(特別な記載がない限り、以後「MDシミュレーション」と記載した場合は採用した手法を指します。) (図2.2)。
いくつかの拡張アンサンブル手法では、事前に構造変化や分子の結合・解離反応等の観察したい任意の反応を表現する方向 (反応座標)を定義しておき、自由エネルギー曲面上をその方向に沿ってランダムウォークさせることにより構造サンプリングを効率化します。しかし反応座標はシミュレーション系や観察対象によって異なる上、その反応座標が本当にその反応を表現しているかどうかが明らかではない等の問題点により、一般に反応座標の決定は容易ではありません。一方でaMD法は反応座標を必要としない拡張アンサンブル手法であり、相対的に利用が容易であることから本検証で使用しました。
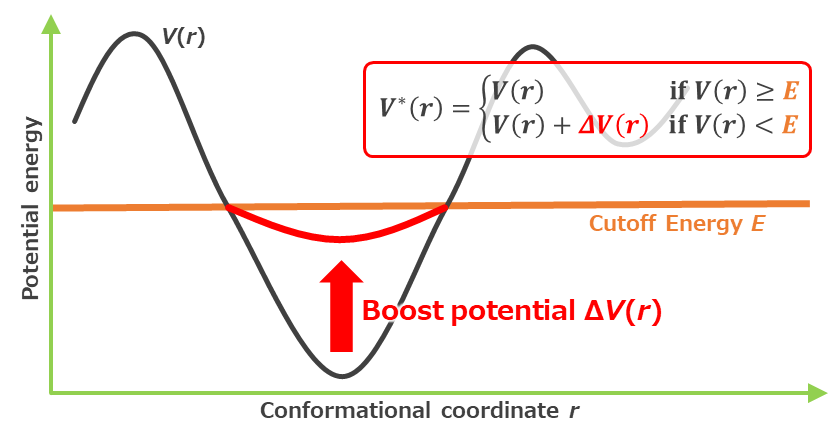
図2.2. Accelerated MD (aMD)法*7の概略。系のポテンシャルエネルギー$V(r)$が閾値$E$よりも低い場合にブーストポテンシャル${\Delta}V(r)$を印加し、系のポテンシャルエネルギー曲面を平滑化することでサンプリング効率を向上させる。
MDシミュレーションによる構造最適化については弊社内で様々な試行錯誤を行っていますが、本検証においては最終的に§1で作成した16個のヒトAhRのクラスター代表構造に対して、弊社にて選定した代表的なantagonistとの複合体構造を作成しMDシミュレーションを実施といったアプローチを採用しました。MDシミュレーションの後半部分から得られたsnapshot構造を解析すると、(ColabFoldのテンプレート構造としても使用した) AhRの既知結晶構造 (apo, holo状態)とは異なる構造状態がサンプリングされたことが分かります (図2.3)。この結果から、大別して5つの準安定状態が存在することが示唆されます。
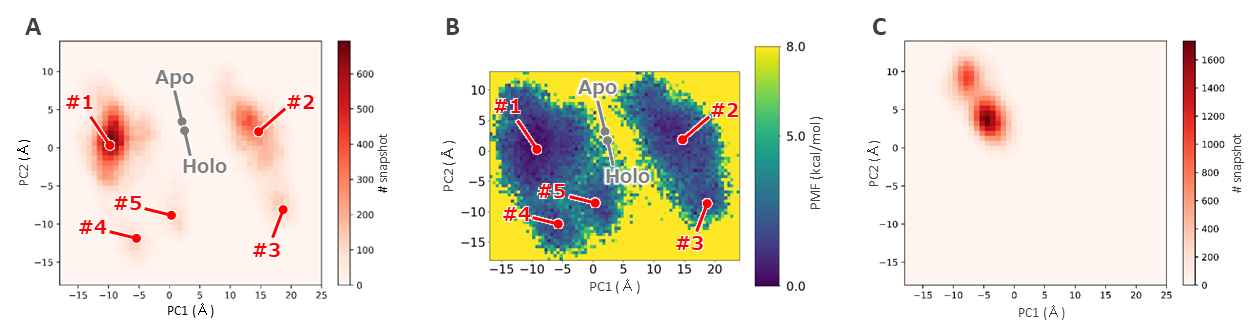
図2.3. MDシミュレーションから得たsnapshot構造の分布。(A) 二次元ヒストグラム、(B) PMF。x, y軸はそれぞれ第一および第二主成分軸。(C) 同時間のclassicalなMDシミュレーションから得たsnapshot構造の分布。本検証にて採用した手法によるMDシミュレーションと比較して、一部の構造状態しかサンプリングされていないことがわかる。
2.2. ドッキングシミュレーションによる最適化構造の評価
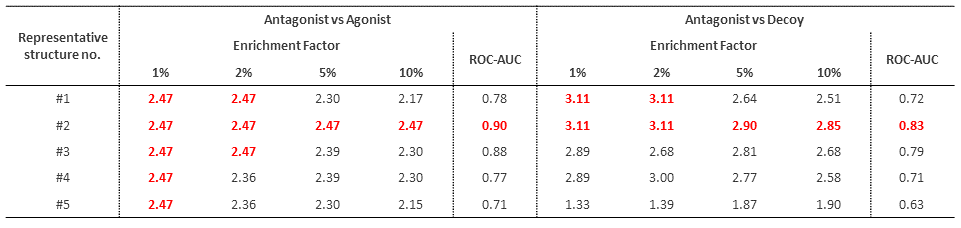
まずMDシミュレーションから得たsnapshot構造を5つのクラスターにグループ分けし、各クラスターの代表構造それぞれについて§1.3と同様のドッキングシミュレーションによりantagonistの予測性能を評価しました。その結果、どの構造についても予測性能が大幅に向上しました (表2.1)。特に予測性能が最も良好だった構造 (#2) のROC-AUCは、従来のホモロジーモデリングとclassicalなMDシミュレーションの組み合わせによる構造のそれ (antagonist vs agonist: 0.80, antagonist vs decoy: 0.79)*2を上回ります。
表2.1. MDシミュレーションから得た5構造のドッキング評価結果
3. まとめと課題
本記事では、弊社事例紹介*2でも紹介しましたヒトAhRに対するantagonistのSBVS探索を対象に、タンパク質立体構造予測ソフトウェアとMDシミュレーションによる構造最適化を組み合わせた構造モデリングの事例をご紹介しました。結論として、今回のヒトAhRについては既知結晶構造の問題 (§1.3)もありColabFoldの予測構造をSBVSにそのまま使用することは困難でしたが、MDシミュレーションを用いた構造最適化を組み合わせることによりSBVSを実施可能とみられる程度の予測性能を有するモデリング構造を得ることが出来ました。
なお本検証におけるヒトAhRのモデリング構造のantagonistの予測性能には従来のホモロジーモデリングとMDシミュレーションの組み合わせから得た構造と大きな違いはありませんが、モデリング構造を得るまでに必要な作業工数に大きな差があったことを特記します。SBVSでは標的タンパク質構造の善し悪しがフロー全体の精度に大きく影響するため、たとえ結晶構造をそのまま使用する場合でもよく注意して構造を準備する必要があります。従来のホモロジーモデリングの場合、モデリング対象のアミノ酸配列とテンプレート構造のアミノ酸配列との配列類似度やその構造の解像度・実験条件といった多くの因子を注意深く検討する必要がありますが、これらは現状では人手による作業が必要な部分が多いため、どうしてもある程度まとまった作業工数が生じます。一方で本検証では、タンパク質立体構造予測ソフトウェアとしてColabFoldを活用したことにより作業の多くの部分をColabFoldで代替することが可能となり、結果として§1のヒトAhRの構造予測に要した作業工数を大幅に削減(約75%減)することが出来ました。現在も様々なタンパク質立体構造予測手法・ソフトウェアが提案されておりますので、これらを(それぞれの有効性や注意点をよく吟味した上で)うまく活用することで作業効率の大幅な改善につながる可能性があります。
しかしながら本記事でご紹介したアプローチにも課題があります。まず(a) ColabFoldの使用法に関して、今回はColabFoldの構造予測パラメータを変化させることでヒトAhRの複数の異なる予測構造を生成しましたが、このアプローチが一般に有効かどうか(正しい構造を維持したまま変化させることができるか、どの程度異なる構造を生成できるか、など)については今後の検証が必要です。次に(b) 今回採用したMDシミュレーションの手法に関して、他の拡張アンサンブル手法よりも秀でた点がありますが、一方で依然としてエネルギー閾値$E$やシミュレーション時間等の任意パラメータが存在するため、得られた結果には恣意性が残る部分があります。状況に応じて適した構造最適化手法が異なるものと考えられますので、MDシミュレーションに基づくアプローチを取る場合にはそれぞれの拡張アンサンブル手法の長所・短所をよく理解・検討したうえで使用する必要があります。最後に(c) holo状態のタンパク質立体構造予測に関して、最近タンパク質-リガンド複合体構造を直接予測する構造予測ソフトウェアが提案されており*8,*9、これらのツールの活用についても検討する価値があるものと思われます (なおこれらツールを使用して複合体構造を予測した場合でも、おそらくMDシミュレーションによる構造最適化を組み合わせることが可能です)。
さいごに
ゼウレカでは、このようなTarget Analysis単独でのご依頼から、Hit Identification、Hit to lead、Lead optimizationに至るまで、皆様の創薬研究をご支援しています。フィジビリティスタディやin silicoアプローチによる有用性の検討を含むご提案までは無償で対応しておりますので、お気軽にご相談ください。今回の様なたんぱくの構造生成のみや、ADMEプロファイルの予測モデルの構築のみといった個別のプロセスにフォーカスしたようなご相談もお受けしております。下記のコンタクトページよりお問い合わせください!
CONTACT | 株式会社ゼウレカ (xeureka.co.jp)
参考文献
- *1: Jumper, et al. (2021) Nature 596:583–589.
- *2: Xeureka Inc., 【事例紹介】Ultra Large Scale Virtual Screening
- *3: del Alamo, et al. (2022) eLife 11:e75751.
- *4: Mirdita, et al. (2022) Nature Methods 19:679-682.
- *5: LocalColabFold, https://github.com/YoshitakaMo/localcolabfold
- *6: Okamoto (2004) J. Mol. Graphics Model. 22:425-439.
- *7: Hamelberg, Mongan, and McCammon (2004) J. Chem. Phys. 120:11919-11929.
- *8: Lu, et al. (2024) Nat. Commun. 15:1071
- *9: Plainer, et al. (2024) OpenReview, https://openreview.net/forum?id=1IaoWBqB6K